
insights
The Good, the Bad, and the Ugly – One Year into the EU Clinical Trial Regulation
Earlier this year, we convened at CORE, Halloran’s Clinical Operations Retreat for Executives, to plan for the future of clinical trials. One of the panels, “Real Life Experience: the Good, the Bad, and the Ugly One Year into the EU Clinical Trial Regulation,” moderated by Halloran’s Mamta Puri-Lechner, reviewed the impact of the new regulation and gathered tales from her panel.
Panelists included Dana Niedzielska, CEO of August Research, Carrie Melvin, SVP, Head of Global Clinical Development Operations at Stemline, Samantha Zappia, VP of Regulatory Affairs and Quality at Vigil Neuroscience, and Danielle Pelletier, Director of Regulatory Affairs at Edgewise Therapeutics.
Before we dive into the one-year assessment, here is a brief background on the European Union Clinical Trial Regulation (EU CTR). As of January 31, 2023, all new drug-based clinical trials must be submitted under the EU CTR with the goal of harmonizing clinical trials across all EU member states. Ongoing studies may continue to remain under the old Clinical Trial Directive (CTD); however, January 31, 2025, is the deadline when all new and ongoing clinical studies must be submitted in accordance with the CTR.
The goal of the regulation is to increase the number of trials in Europe, and considering EU states were all working separately, this new regulation will centralize and streamline the clinical trial startup process.
Though many sponsors are still in flux, we anticipate more centralization and streamlining in the next couple of years. Here is a bite-sized overview of the pros and cons, and a few challenges experienced along the way.
The Good Side of the EU CTR
The EU CTR offers many positives, including:
- Harmonized process across all EU member states
- Single platform for submission with one application to all member states, resulting in a lack duplicated efforts
- Strict timelines for submission and review processes across all member states
- Collaborated assessment and decision making leading to a reduction in country-specific activities and documents
- Reduced number of translations and other country-specific activities leading to a streamlined process
- Reduced number of information requests due to a collaborative assessment
- Enhanced transparency of clinical trial data
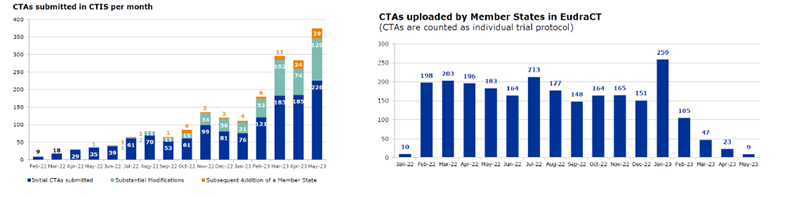
As of May 2023, the number of initial Clinical Trial Applications (CTAs) submitted in accordance with the EU CTR are increasing, alongside the number of subsequent additional member states. When compared to EudraCT, the previous platform, we see the EudraCT numbers drop significantly, as expected. See Figure 1.
Figure 1
Navigating Bad Aspects of the EU CTR Process
As EU clinical trial sponsors adapt to the new regulation, some find the experience not quite easy and straight forward to navigate. Here is a tip; One of the first decisions a sponsor will need to make is that of the Reporting Member State (RMS) for the application.
The RMS takes ownership of the application, including the coordinated assessment and harmonized conclusion of the process, and hence, serves as a point of contact between the sponsor and the member states involved in the application. The role of the RMS is critical and must not be taken lightly but should also not be overstated. Each member state gets to have its own independent assessment of the application, and the RMS role is to collate these reviews and correspond with the sponsor. This process is not bad per say, it is simply different. Sponsors will need to prepare and plan accordingly.
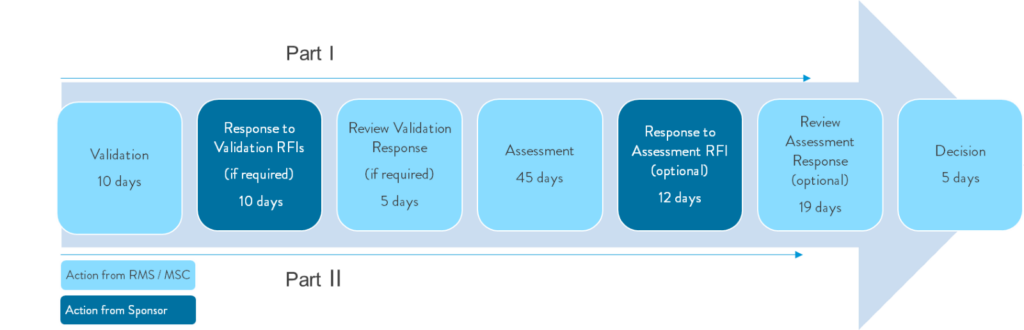
Another challenge sponsors face when adapting to the EU CTR is the timeline for the assessment of the application. This new timeline includes a short turnaround time of 12 business days for the sponsor to respond to any Requests for Information (RFI). This means all sponsors will need to be prepared to respond to all incoming communication in a timely manner, and this will require a focused team readily available during the assessment phase.
It is important to keep in mind that you may need to hire to prepare for the change in timeline and expectations. A panelist noted, “there are not cost savings with this development. Now, you need a larger team to move through the process.” Consider your current resources and do an analysis to see if what you have will get you to where you need to be. This new process is regimented so you do not have the flexibility with deadlines.
If you are unfamiliar with the timeline, the figure below shows the parallel submission and review process for both parts of the application. Part 1 is the common application, and Part 2 is the member state specific application, and both parts need to be incorporated into the planning process for a submission via the EU CTR.
Figure 2
Has the Past Year Been Ugly? A Look into Industry Response
While the good aspects of the EU CTR will be a streamlined and centralized regulatory submission process, some sponsors are facing challenges.
One year into the regulation, a few challenges were shared during the panel discussion. One panelist acknowledged, “The transition is difficult because the new CTR has different regulations than the CTD, and many sponsors are feeling some challenges, like around Informed Consent Form (ICF) rules in the Ethics Review.” Considering this regulation also aims to uphold higher standards for patient safety and to increase transparency, patient-centric provisions for ICF have been added. Under Article 29 in the EU CTR, improvements and expansions of definitions of clinical trial participation and guidelines for consent and involvement have been introduced.
“There will likely be time savings on Part 1 of the process, but on the ethics side, you still have to translate ICFs, and there are many details to consider in the process,” noted another panelist.
In addition, the EU CTR asks for all commercially confidential information to be redacted. Yet, sponsors do not know what information should or should not be redacted. And sponsors certainly should not over redact.
Navigating this new regulation is complex, requiring a different approach going forward when submitting CTAs in EU member states. With a little over a year to go when all clinical trials must be submitted in accordance with the CTR, now is the time to plan and prepare.
As you navigate your EU CTR submissions, Connect with Halloran to discuss your pressing regulatory operations questions and concerns. We are ready when you are.
References: