
insights
Is the Accelerated Approval Pathway at Risk?
The health care industry is still reeling from the aftershocks of the accelerated approval of Biogen’s Alzheimer’s drug, aducanumab. From reimbursement and health care coverage woes to Acting FDA Commissioner Janet Woodcock requesting an investigation into the approval of aducanumab, events are still unfolding. Unfortunately, and not surprisingly, the accelerated approval pathway has come under intense scrutiny. The US Office of Inspector General (OIG) is expected to provide a report in 2023 with an assessment of how the FDA implements the accelerated approval pathway.
Rick Pazdur, MD, FDA’s Head of Oncology Center of Excellence (OCE), a strong advocate for the accelerated approval pathway has stated he feels the accelerated approval pathway has come “under attack” following the approval of aducanumab. Dr. Pazdur has firsthand experienced the loss of a loved one from cancer. He understands the importance, value, and hope for patients who may have exhausted all available treatment options and are desperate to try a new therapy that was approved based on surrogate endpoints rather than definitive effects on overall survival.
The accelerated approval pathway allows the FDA to approve drugs that treat a serious condition, provide a meaningful advantage over available therapies, and demonstrate an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The magnitude of improvement over available therapy required to warrant accelerated approval is based on a benefit and risk analysis of the totality of evidence and is an NDA/BLA review issue.
Following accelerated approval, a confirmatory trial is required to confirm the clinical benefit (e.g., effects on overall survival) and then the study results are reported back to the FDA to convert the accelerated approval to full approval. FDA repeatedly reminds sponsors that a confirmatory study must be well underway, if not fully enrolled, at the time of an accelerated approval action. If sponsors don’t follow through on this mandate, FDA could halt product approval or, in cases of a negative confirmatory study, the FDA would rescind the accelerated approval.
The accelerated approval pathway is used most frequently for oncology and immuno-oncology therapies. Since the implementation of accelerated approval, many drugs have been approved under this regulatory pathway. Thanks to significant scientific innovation made by our industry, the number of drugs approved under the accelerated approval pathway has skyrocketed within the last two years. See the chart below from STAT News.
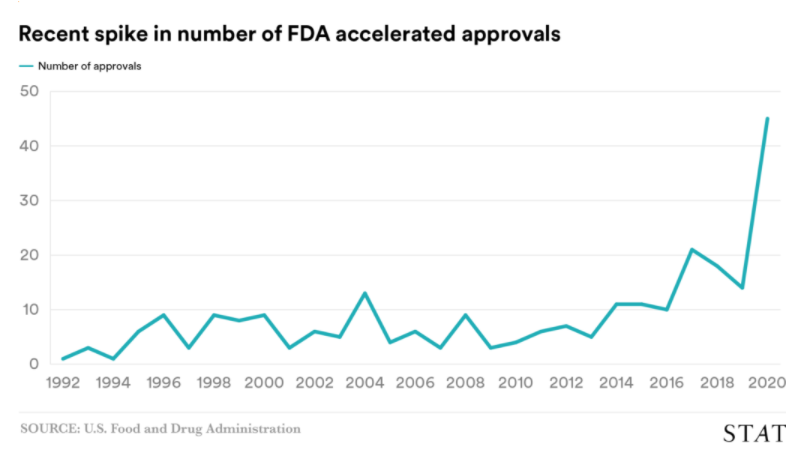
Although we don’t yet know the outcome of the OIG investigation, or how the outcome will impact the accelerated approval pathway moving forward, we are already seeing its effects. More and more sponsor company press releases are reporting the FDA issuing Complete Response Letters (CRLs) to sponsors requesting additional clinical safety and efficacy data, and additional randomized, controlled clinical trials in order to approve new therapies under the accelerated approval pathway.
In Halloran’s recent experience, the scrutiny of accelerated approval was clearly on display during an FDA mid-cycle communication meeting for the BLA review of immuno-oncology therapy. The FDA was critical of “marginal efficacy” even though the study results were comparable to other approved immuno-oncology therapies in the same drug class. Given the recent discussions at the Oncologic Drugs Advisory Committees (ODACs) dated April 27-29, 2021, and June 24, 2021, the FDA believes it’s unclear that the magnitude of Overall Response Rate (ORR) from a single-arm trial is likely to predict clinical long-term benefit and is not regarded favorably. The FDA was also critical of whether the confirmatory trial would be able to fully enroll and confirm the benefit in a reasonable timeframe.
During the BLA safety review of the same immuno-oncology therapy noted above, the FDA identified certain categories with higher rates of serious adverse events (SAEs) compared to other available therapies and potentially more interruptions from immune-related events. However, SAE differences are variable across studies, institutions, and practice patterns as the decision to admit a patient for toxicity management is clinician dependent (admission to hospital alone is a defining SAE event).
SAEs are common in advanced cancer trials and are typically related to disease progression, not study drugs. Small numerical differences with no statistical significance between SAEs across trials are not clinically meaningful. There are pitfalls of cross-study comparison – it is challenging to compare efficacy across clinical trials, and it is equally challenging to compare safety between drugs and trials.
Although the accelerated approval pathway may seem like it is at risk, our team at Halloran does not see this pathway dissipating. This regulatory pathway has a tremendous impact on the lives of countless patients and removing this pathway as an outcome of the OIG investigation seems unrealistic. It would be a major step backward in bringing new therapies to patients as quickly as possible to address an unmet medical need. However, our team envisions more conversations and meetings with the Agency about the applicability of the accelerated approval pathway to a drug program and the data needed to support an accelerated approval to ensure clinical benchmarks are met.



